This web page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison .
What is phylogeny?_________________________________________________________________________________________
Phylogeny concerns the evolutionary relationships between species and how closely related they are.
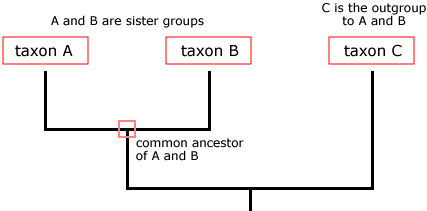
Phylogenetic trees provide visual representations of these relationships: As summarized in the diagram below, the tips of the trees each represent one species, and a node represents a common ancestor from which the species at the tips descended. When different species (or daughter lineages) are descended from the same common ancestor, they are known as sister groups. Sister groups are always the same age since they arose from the same ancestor at approximately the same time. The outgroup is the species that is not as closely related to members of a group of interest. The branch lengths of a tree represent the number of nucleotide substitutions divided by the length of a sequence. The longer the branch, the higher the number of changes in the organism's genome in relation to another organism's genome in the tree [1].
Phylogeny concerns the evolutionary relationships between species and how closely related they are.
Phylogenetic trees provide visual representations of these relationships: As summarized in the diagram below, the tips of the trees each represent one species, and a node represents a common ancestor from which the species at the tips descended. When different species (or daughter lineages) are descended from the same common ancestor, they are known as sister groups. Sister groups are always the same age since they arose from the same ancestor at approximately the same time. The outgroup is the species that is not as closely related to members of a group of interest. The branch lengths of a tree represent the number of nucleotide substitutions divided by the length of a sequence. The longer the branch, the higher the number of changes in the organism's genome in relation to another organism's genome in the tree [1].
How are the trees generated?_______________________________________________________________________________
First, similarity scores are calculated. This is achieved by either the BLOSUM matrix or percentage identity.
A BLOSUM matrix is used to score alignments between pairs of protein sequences. The numbers in BLOSUM matrices are based on the different probabilities of each nucleotide aligning. Jalview--a plugin software that allows the visualisation and analysis of alignments--uses the BLOSUM62 matrix, which can be viewed here. For generating a tree, a score is assigned to each amino acid pair. The scores are then added up. The higher the score, the more closely related the sequences are.
The percentage identity method involves what percentage two sequences are identical to each other. It is essentially the number of equivalent aligned amino acids per 100 amino acids [2].
An average distance tree assumes that the rate of evolution is constant across lineages. A distance matrix with similarity scores representing how closely related the species are is used to generate the tree [3].
In generating a neighbour joining tree, an algorithm to generate a tree with the shortest possible branch lengths is applied.
The following trees for comparing DRD2 gene sequence similarity between species were generated:
First, similarity scores are calculated. This is achieved by either the BLOSUM matrix or percentage identity.
A BLOSUM matrix is used to score alignments between pairs of protein sequences. The numbers in BLOSUM matrices are based on the different probabilities of each nucleotide aligning. Jalview--a plugin software that allows the visualisation and analysis of alignments--uses the BLOSUM62 matrix, which can be viewed here. For generating a tree, a score is assigned to each amino acid pair. The scores are then added up. The higher the score, the more closely related the sequences are.
The percentage identity method involves what percentage two sequences are identical to each other. It is essentially the number of equivalent aligned amino acids per 100 amino acids [2].
An average distance tree assumes that the rate of evolution is constant across lineages. A distance matrix with similarity scores representing how closely related the species are is used to generate the tree [3].
In generating a neighbour joining tree, an algorithm to generate a tree with the shortest possible branch lengths is applied.
The following trees for comparing DRD2 gene sequence similarity between species were generated:
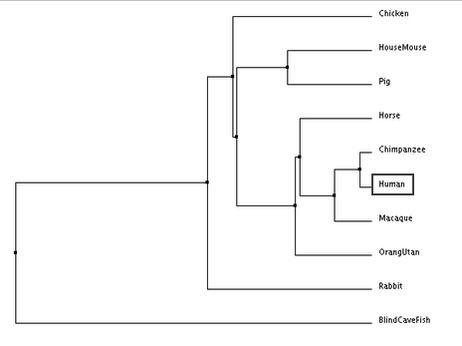
Average distance tree using % identity
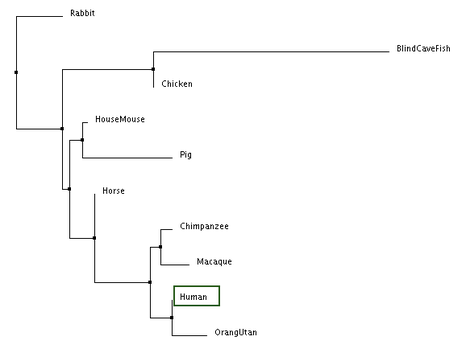
Neighbour joining tree using % identity
|
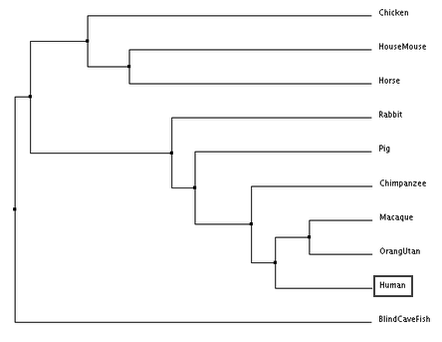
Average distance tree using BLOSUM62
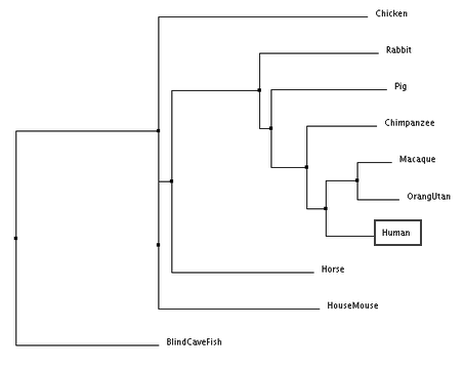
Neighbour joining tree using BLOSUM62
|
Discussion__________________________________________________________________________________________________
According to all four trees, the blind cave fish DRD2 gene sequence is the most distantly related compared to humans. In the trees generated, humans and primates (chimpanzee, macaque and orang utan) are consistently seen to be closely related as they are grouped together in one clade. The results generated are similar to those obtained from generating phylogenetic trees using DRD2 protein sequences. However, it is interesting to note that despite chimpanzee DRD2 gene sequences being the most closely related to the human sequence, chimpanzees seem to be the least closely related clade member to humans relative to other primates, with the exception of the average distance tree generated using percentage identity.
According to all four trees, the blind cave fish DRD2 gene sequence is the most distantly related compared to humans. In the trees generated, humans and primates (chimpanzee, macaque and orang utan) are consistently seen to be closely related as they are grouped together in one clade. The results generated are similar to those obtained from generating phylogenetic trees using DRD2 protein sequences. However, it is interesting to note that despite chimpanzee DRD2 gene sequences being the most closely related to the human sequence, chimpanzees seem to be the least closely related clade member to humans relative to other primates, with the exception of the average distance tree generated using percentage identity.
References
[1] Reading trees: A quick review. http://evolution.berkeley.edu/evolibrary/article/phylogenetics_02
[2] Calculation of trees from alignments. http://www.jalview.org/help/html/calculations/tree.html
[3] Phylogenetic Reconstruction. http://evolution-textbook.org/content/free/contents/ch27.html#ch27-4-2

{kind=link}